
MUC1通过稳定EGFRvIII促进胶质母细胞瘤进展和TMZ耐药

表皮生长因子受体Ⅲ型突变体(EGFRvIII)缺失外显子2-7,其稳定性的机制尚不清楚。基于CRISPR-Cas9文库筛选,天津医科大学总医院王琦雪和康春生课题组发现MUC1对EGFRvIII胶质瘤细胞存活和替莫唑胺(TMZ)耐药性至关重要。他们发现MUC1-C在EGFRvIII阳性细胞中上调并增强了EGFRvIII的稳定性。MUC1-C的敲除增加了EGFRvIII和溶酶体的共定位。MUC1的上调以NF-κB依赖的方式发生,抑制NF-κB途径可以通过抑制MUC1-C来中断EGFRvIII-MC1反馈回路。在之前的研究中,课题组发现了AC1Q3QWB(AQB),AQB是一种可以抑制NF-κB磷酸化的小分子。通过筛选AQB的结构类似物,发现了EPIC-1027,它可以更有效地抑制NF-κB通路。EPIC-1027在体外和体内破坏了EGFRvIII-MUC1-C正反馈回路,抑制了胶质瘤的进展,并促进了对TMZ的敏感性。总之,本研究揭示了MUC1-C在胶质母细胞瘤(GBM)中稳定EGFRvIII的关键作用,并发现了一种小分子EPIC-1027,在GBM治疗中具有巨大潜力。
胶质母细胞瘤(GBM)是成人最恶性的原发性颅内肿瘤。GBM患者的标准护理是手术切除,然后联合替莫唑胺(TMZ,胶质瘤的一线化疗药物)和放射治疗。然而,该治疗在预防GBM的恶性进展和提高患者生存率方面无效。EGFR是GBM中最常见的突变基因,EGFR突变体III(EGFRvIII)是常见的恶性基因组突变。EGFRvIII中外显子2–7的缺失从受体的胞外结构域中删除了267个氨基酸。尽管EGFRvIII不能结合配体,但它具有组成性活性。与野生型EGFR类似,EGFRvIII可以激活RAS/MAPK、PI3K/AKT和JAK/STAT通路。然而,与EGFR相比,EGFRvIII阳性胶质瘤细胞在DNA复制、DNA修复和血管生成途径中高度富集。EGFRvIII+和EGFRvIII– GBM细胞的转录本出现显著性表达差异,CDK4和MDM2扩增在EGFRvIII+患者中发生频率较低。EGFRvIII+胶质瘤细胞可以通过分泌生长因子重塑肿瘤微环境,从而促进GBM进展。据报道,EGFRvIII也转移到细胞核中,以促进放疗和化疗期间的DNA修复,抑制缺氧诱导的细胞凋亡。EGFRvIII被认为是一种稳定的蛋白质,易于逃避内吞和降解。Kim等人报道ANO1可以稳定EGFRvIII的表达,并保持GBM干细胞的干性。尽管CBL蛋白和LRG1已被报道介导EGFRvIII的下调,但GBM中EGFRvIII调节的靶位点仍有待确定。
黏蛋白1(MUC1)是一种跨膜糖蛋白,在多种上皮癌中过度表达。MUC1基因编码单个多肽链,由于构象应力,该多肽链被自催化降解为较长的N末端亚基(MUC1-N)和较短的C末端亚单位(MUC1-C)。尽管有关于MUC1-N在肿瘤进展和化疗耐药中的作用的报道,但MUC1-C在癌症进展中的作用已引起越来越多的关注。基于糖基化状态,MUC1-C的分子量可以在17-25kDa之间变化。MUC1-C的异常表达促进肿瘤进展,重新编程癌症代谢,介导化学耐药,维持细胞干性,并改写肿瘤细胞表观遗传学。然而,MUC1在GBM中的作用尚不清楚。
PRC2复合物可以介导组蛋白H3K27的甲基化,并改变细胞染色质的表观遗传修饰。因此,PRC2组分(EZH2、SUC12和EED)的异常表达通常与癌症的进展和不良预后相关。康春生教授课题组团队鉴定了两种小分子化合物,AC1NOD4Q(ADQ)和AC1Q3QWB(AQB),通过特异性阻断lncRNA HOTIAR对PRC2的募集,用于治疗胶质瘤和乳腺癌。AQB可以通过表观遗传学抑制H3K27me3-UBXN1-NF-κB通路来抑制PD-L1表达并逆转胶质瘤中的免疫逃逸。小分子抑制剂由于其结构简单、易于合成和高效,是特别具有潜力的治疗策略。
本研究中,我们基于CRISPR-Cas9文库和CGGA原发性和复发性胶质瘤数据库,在EGFRvIII阳性胶质瘤细胞中发现MUC1为TMZ抗性和生存相关基因。MUC1的过度表达促进了体外和体内EGFRvIII胶质瘤细胞的增殖。MUC1不影响EGFRvIII的mRNA水平,但通过自噬相关信号通路稳定其蛋白水平。此外,我们筛选了EPIC-1027作为AQB的更好衍生物,它提高了GBM细胞和颅内胶质瘤模型中TMZ的敏感性,显示出巨大的治疗性应用潜力。
1.MUC1促进EGFRvIII阳性胶质母细胞瘤细胞的增殖和TMZ抗性

此前,我们使用CRISPR/Cas9文库研究U87MG/U87 EGFRvIII细胞中的TMZ抗性基因(图1A)和胶质瘤细胞中对TMZ抗性至关重要的191个基因(图1B)[33]。单变量Cox分析显示18个基因与原发性和复发性胶质瘤患者的无进展生存率(PFS)高度相关;最显著相关的p值基因是MUC1(图1C)。患者信息如表1所示。当比较第14天U87vIII和U87MG的sgRNA-seq数据时,我们发现U87vII细胞中57个编码基因的sgRNA显著减少,表明这些基因是EGFRvIII的潜在致死基因(图1D)。MUC1是TMZ抗性和EGFRvIII生存相关基因列表中唯一存在的基因,表明MUC1在EGFRvIII-阳性胶质瘤细胞中发挥重要作用(图1E)。生存分析表明,MUC1的高表达与CGGA原发性(325例患者;85例原发性GBM)和复发性GBM(693例患者;104例复发性GBM)的不良预后相关(图1F)。生物信息学分析显示,MUC1 mRNA水平在IV级胶质瘤中最高,并在GBM的间充质亚型中富集(图1 G,H)。与MUC1高度相关的基因在上皮细胞增殖、ERK1/2、JAK-STAT和PI3K-AKT途径中富集,表明MUC1参与EGFR功能(图1I)

2.MUC1增强了EGFRvIII蛋白的稳定性

TBD0220是具有天然EGFRvIII突变的原代培养GBM细胞系,通过western blotting和RT-PCR对其进行了分析(图2A-B)。使用特异性抗体检查MUC1-N或MUC1-C表达(图2C)。TBD0220细胞中MUC1的过度表达促进了增殖,而shRNA敲低MUC1抑制了细胞生长速度(图2D,E)。MUC1表达的变化可以以类似的方式影响U87vIII细胞增殖(图2 F,G)。我们用EGFRvIII和MUC1-C过表达慢病毒转染U87、LN229和N33细胞。MUC1的过度表达增强了wt EGFR的表达,而MUC1的敲低降低了U87和LN229细胞中外源性EGFRvIII的表达(图2H)。Western blot分析表明,EGFRvIII转染增加了MUC1-C而非MUC1-N蛋白水平,而MUC1-C也增加了外源EGFRvII表达(图2I)。TBD0220细胞表达内源性EGFRvIII,western blotting表明shRNA敲低MUC1降低了EGFRvII的表达(图2 J)。从这些结果中,我们得出结论,EGFRvIII可以上调MUC1-C蛋白水平,而MUC1-C可以增强外源和内源性EGFRvⅢ的表达。
为了研究MUC1-C如何调节EGFRvIII表达,我们检测了MUC1过表达或敲低后内源性EGFRvIIImRNA水平(图2 K,L)。EGFRvIII mRNA水平随MUC1水平的变化几乎没有变化,表明转录后调节(图2M)。蛋白质稳定性分析表明,shMUC1加速了内源性和外源性EGFRvIII和EGFR蛋白质的降解(图2N-Q)。此外,EGFRvIII不能影响MUC1的mRNA水平(图S1A),但增强了MUC1-C蛋白的蛋白质稳定性(图S1B),这意味着它们在转录后水平上相互调节。
为了研究MUC1-C稳定EGFRvIII的机制,我们用MG132(蛋白酶体抑制剂)和氯喹(CQ,自噬抑制剂)处理U87vIII-shMUC1和TBD-shMUC1细胞。Western blot分析显示,shMUC1显著降低EGFRvIII表达,而CQ处理在24小时部分逆转了这种抑制(图3A,B)。有趣的是,MG132显著降低了EGFRvIII蛋白水平。我们假设这可能与10μM剂量的MG132的细胞毒性有关。在用MG132以0.1、0.5和1μM重复测试后,我们发现EGFRvIII仍然被MG132抑制,但被CQ上调(图3 C,D)。

我们将注意力转移到CQ和自噬途径的影响上。共聚焦成像分析显示,shMUC1引导的EGFRvIII与溶胞蛋白酶抑制剂(溶酶体的标志物)在TBD0220细胞中共定位。在CQ治疗下,很少发现EGFRvIII和赖氨酸受体拮抗剂的共定位,以及增强的EGFRvII表达(图3E)。

在我们用MUC1-C、EGFRvIII和EGFRvIII/MUC1-C共转染的U87MG细胞构建的颅内胶质瘤模型中(图4A),我们发现EGFRvII/MUC1-C显著促进了体内胶质瘤的进展(图4B~D)。IHC分析表明,在EGFRvIII/MUC1-C共转染的GBM样品中,Ki-67和EGFRvIII的染色增强,肿瘤边缘浸润增加(图4E,F)。
3.靶向NF-κB通路可阻断EGFRvIII/MUC1-C的自稳定
我们旨在表征EGFRvIII/MUC1-C用于GBM治疗的自我稳定机制。生物信息学分析揭示了MUC1启动子中的NF-κB结合位点(图5A)。将NF-κB抑制剂JSH-23-添加到TBD0220细胞中,MUC1启动子的活性降低。然而,当NF-κB结合位点突变时,抑制作用消失(图5B)。然后将JSH-23添加到TBD0220(C)和U87vIII(D)细胞中,实时PCR分析显示MUC1 mRNA受到抑制。Western blot分析证实了JSH-23对MUC1-C表达的抑制作用(图5E)。

AQB是一种小分子,可以抑制HOTAIR和EZH2的相互作用,并阻止PRC2复合物进入细胞核[25]。通过该功能,AQB阻止H3K27的三甲基化,并通过降低启动子组蛋白的甲基化水平释放p21和UBXN1的表达。UBXN1是NF-κB的抑制因子,AQB通过上调UBXN1抑制NF-κB的磷酸化。因此,AQB可以通过H3K27me3-UBXN1-NF-κB轴抑制胶质瘤中的p-NF-κB。
我们进行了下一代小分子化合物设计和合成,发现EPIC-1027与AQB具有结构和功能上的相似性,在物理上形成了HOTAIR和EZH2之间的嵌合体(图5 F,G)。CCK8分析显示EPIC-1027的IC50显著低于AQB(图5H,I)。FACS分析和蛋白质印迹也显示EPIC-1027具有更好的细胞周期阻滞作用(图S2A,B)。作为AQB的衍生物,EPIC-1027自然地被测试对H3K27me3和p-NF-κB的抑制作用。实时PCR分析显示,EPIC-1027可以剂量依赖性方式上调UBXN1 mRNA水平,类似于AQB(图5 J,K)。使用相似浓度的AQB和EPIC-1027,我们发现TBD0220和U87vIII细胞在用EPIC-10270处理时对H3K27me3和p-NF-κB的抑制作用更显著(图5 L,M)。EPIC-1027可降低MUC1启动子的活性;然而,当NF-κB结合位点突变时,抑制作用消失(图5N)。一致地,EPIC-1027以剂量依赖性方式抑制TBD0220和U87vIII细胞中的MUC1 mRNA水平(图5 O,图S2C)。NF-κB负责EGFRvIII相关的TMZ抵抗,并在TMZ治疗下被激活[33]。我们用TMZ和EPIC-1027处理EGFRvIII阳性胶质瘤细胞。实时PCR分析显示,TMZ处理上调了MUC1,而EPIC-10270逆转了这种上调(图5 P,图S2D)。EPIC-1027抑制MUC1表达的机制如图所示。5Q。
MUC1是TMZ抗性基因(图1C),并降低了EGFRvIII阳性细胞中TMZ的IC50(图6A,B)。共焦成像分析显示MUC1的过度表达减少了γ-H2A的数量。TMZ治疗后的x个病灶。同时,MUC1的敲除增强了TMZ诱导的细胞凋亡(图6C)。Western blot分析显示,shRNA敲低MUC1抑制了TMZ处理的细胞中p-CHK2、RAD50和E2F1的表达,它们在DNA损伤修复中具有重要功能(图6D)。总之,这些结果揭示了MUC1在EGFRvIII阳性GBM细胞TMZ耐药性中的重要作用。
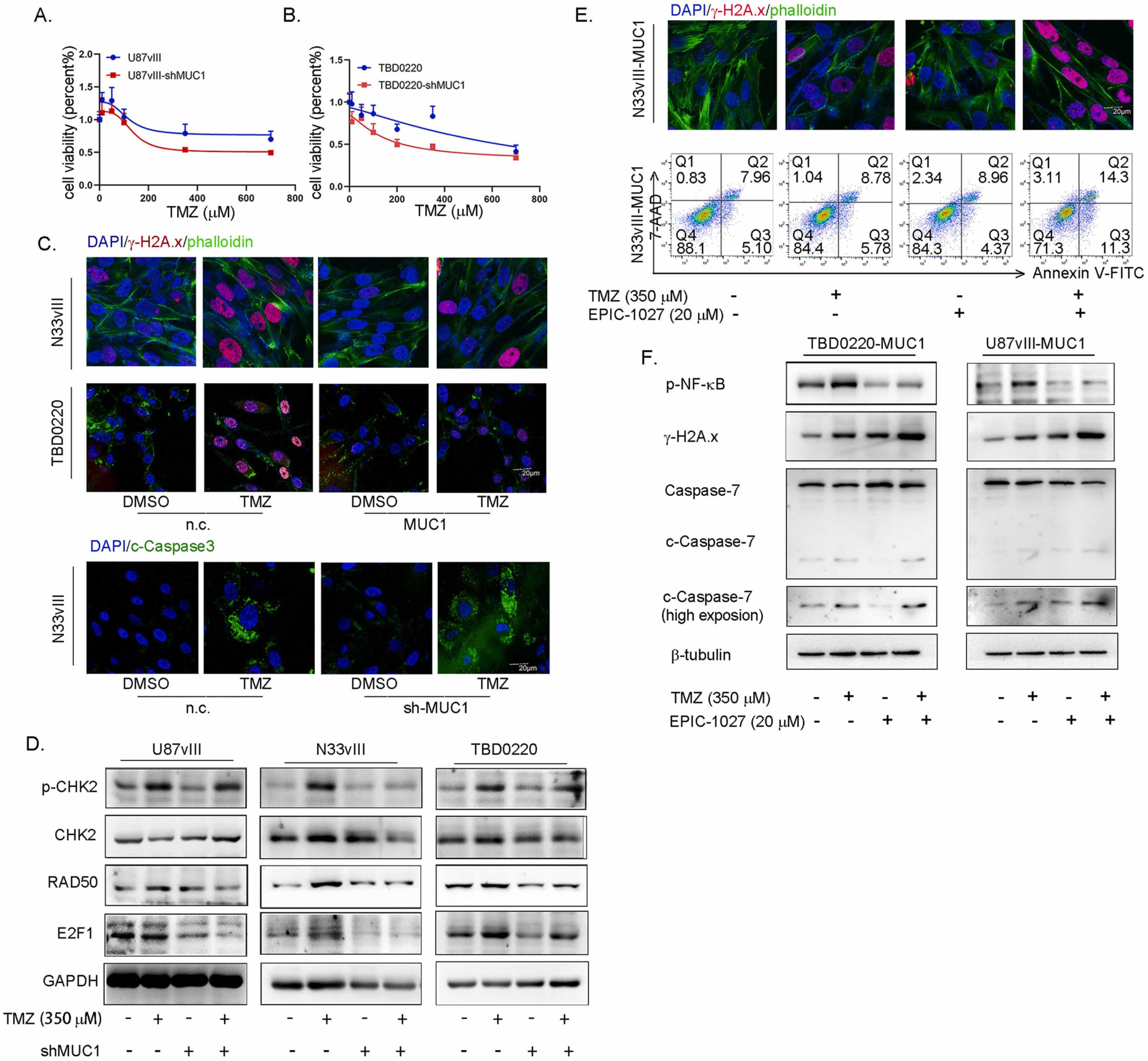
因此,我们测试了TMZ在EGFRvIII-MC1阳性胶质瘤细胞中的增敏作用。联合治疗后,共聚焦分析显示γ-H2A的数量增加。x病灶,而FACS分析表明,在N33vIII-MC1细胞中,TMZ和EPIC-1027共处理组中凋亡率较高(图6E)。Western blot分析显示,联合治疗增强了DNA损伤和凋亡,表明EPIC-1027增强了TBD0220-MUC1和U87vIII-MC1细胞对TMZ的敏感性(图6 F)。
最后,我们在体内测试了EPIC-1027的肿瘤抑制作用(图7A)。MRI图像表明EPIC-1027显著增强了TMZ对颅内胶质瘤进展的抑制作用(图7B)。组合治疗也延长了生存时间(图7 C)。IHC分析显示联合治疗组p-NF-κB和Ki-67表达减少(图7D)。有趣的是,在TMZ治疗后,EGFRvIII在细胞核中染色,这与EGFRvII可以在化疗和放疗下协助DNA损伤修复的报道一致[8]。EPIC-1027治疗逆转了这种表型(图7D)。
为了评估EPIC-1027的血脑屏障穿越能力,我们用TBD0220细胞构建了颅内胶质瘤模型,并分析了药物分布。给药EPIC-1027(7.5mg/kg)后24小时处死小鼠。收集脑、肾、肺、脾、肝和心脏组织。EPIC-1027的含量通过液相色谱-三重四极质谱法进行分析,并列于表S1中。结果表明,在大脑中存在低但可检测到的EPIC-10270分布,肿瘤承载侧的药物含量略高。因此,我们确认了EPIC-1027在脑组织中的存在。

为了评估EPIC-1027的生物安全性,c57bj小鼠口服给药7.5 mg/kg,持续两周。我们的生化测试和H&E染色表明EPIC-1027具有生物安全性(表S2、S3和图S3),并增强了体外和体内对化疗的敏感性。因此,这些结果表明EPIC-1027在胶质瘤治疗中具有良好的应用潜力。
因此,我们得出结论,EPIC-1027增强了体外和体内对化疗的敏感性,具有良好的应用前景
4. 讨论
在这项研究中,我们证明MUC1在EGFRvIII-胶质瘤细胞存活和TMZ抵抗中起重要作用。据我们所知,这是首次揭示MUC1在胶质瘤TMZ抵抗中的作用的研究。MUC1的过度表达降低了DNA损伤的程度,而MUC1的敲除抑制了TMZ治疗下的DNA损伤修复基因,如E2F1。我们研究了MUC1在TMZ抗性中的作用,发现MUC1的过度表达降低了DNA损伤的程度,而MUC1的敲低通过降低DSB修复蛋白(如E2F1)的表达提高了TMZ敏感性。E2F1是参与细胞周期进展和DNA修复的关键转录因子,它介导了胶质瘤中的TMZ抗性。此外,MUC1-C显示与E2F1相互作用,增强其转录活性。因此,我们表明MUC1通过上调E2F1和增强E2F1调节的DDR基因来促进TMZ抗性。
在我们的研究中,我们发现MUC1敲低降低了EGFRvIII的表达,并缩短了CHX治疗下EGFRvII蛋白的半衰期。有趣的是,MG132对胶质瘤细胞的治疗降低了EGFRvIII的表达,即使在非常低的剂量下也是如此。这可能是由于MG132的细胞毒性,据报道,MG132可诱导癌细胞凋亡。共聚焦显微镜显示MUC1敲除促进了EGFRvIII和溶酶体的共定位,表明增强了自噬降解。如在CRISPR-Cas9文库中发现的,MUC1-sgRNA转染显著降低了EGFRvIII阳性细胞的细胞存活率。MUC1的过度表达也会增加野生型EGFR的表达,这与先前的报告一致,其中MUC1改变了EGFR的运输,并阻止了其在乳腺癌中EGF激活下的降解。因此,我们假设MUC1可能在蛋白质稳定性之外发挥重要作用,这将在未来的研究中探索。
我们发现MUC1-C上调EGFRvIII蛋白水平,EGFRvIII上调MUC1-C表达。然而,MUC1的N端在EGFRvIII阴性和阳性细胞之间的表达没有显著变化。此外,EGFRvIII几乎不影响MUC1 mRNA水平,这表明EGFRvⅢ可能促进MUC1的自切割或增强MUC1-C的蛋白质稳定性。生物信息学分析揭示了MUC1启动子上的RELA结合位点,实时PCR和双荧光素酶报告子分析进一步证实了这一点。与我们之前的研究结果一致,我们认为NF-κB抑制可能是EGFRvIII胶质瘤进展和TMZ抵抗的一个有前途的治疗靶点。
EPIC-1027-AQB的衍生物改善了H3K27me3和NF-κB的抑制。我们测试了EPIC-1027对MUC1的抑制作用,并发现MUC1的启动子活性和mRNA水平都受到该分子抑制剂的抑制。此外,TMZ转录上调MUC1,这可以通过EPIC-1027联合治疗逆转。
EPIC-1027和TMZ的联合治疗增强了胶质瘤细胞中的DSB和凋亡,进一步证实了靶向EGFRvIII-MUC1正反馈环在GBM抑制中的有效性。我们将这一治疗方案扩展到小鼠模型,EPIC-1027显著减少了肿瘤体积并延长了生存时间。联合治疗可抑制细胞增殖(Ki-67)和NF-κB通路(p-NF-κ。此外,我们观察到TMZ治疗下EGFRvIII的细胞核易位,据报道,这有助于顺铂和电离辐射治疗后的DNA修复[8]。EPIC-1027取消了这种招募,从而破坏了EGFRvIII-MC1环。最后,我们评估了EPIC-1027的安全性,生化测试表明该分子几乎不会影响组织和器官的正常功能。H&E染色进一步显示了EPIC-1027在小鼠中的健康和完整的组织形态。因此,我们在体外和体内证实了EPIC-1027的抑制作用和TMZ敏化作用。
5.结论
我们的研究表明,MUC1可以稳定胶质瘤细胞中的EGFRvIII,小分子EPIC-1027对MUC1的抑制可以有效抑制肿瘤生长并增强EGFRvIII胶质瘤中的TMZ敏感性。
文献来源:
https://doi.org/10.1016/j.phrs.2022.106606
本研究中MUC1 shRNA慢病毒以及MUC1启动子质粒构建由上海艾博思生物科技有限公司完成

公司网址:www.ibsbio.com 电话:021-38770863


锐竞平台